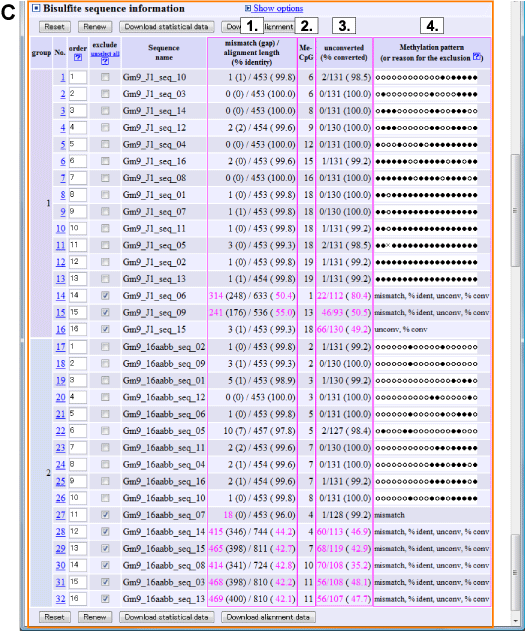
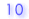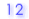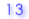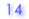| C. Information and methylation pattern of each bisulfite sequences: Two sequence groups are indicated separately. |
 |
| 1. |
Number of mismatches and percent identity of bisulfite alignment |
| 2. |
Number of methylated CpG sites |
| 3. |
Number of bisulfite unconverted CpHs (CpA, CpC, CpT) |
| 4. |
Pattern of CpG methylation (Black circle: methylated, White circle: unmethylated, Cross: mismatch or gap) |
| Methylation pattern (4.) is not shown when quality of bisulfite sequence is low. Low quality value is shown as magenta.
When excluded, reason(s) for the exclusion will be indicated at methylation pattern column (4.).
Conditions to exclude low quality bisulfite sequences can be changed
(See "Show options" for more detail).
|
|
 |
mismatch:
the number of alignment mismatches (includes gaps) between genomic and bisulfite sequences exceeded the upper limit (default: 10).
This means low quality sequence read. |
| |
% ident:
percent of alignment identity between genomic and bisulfite sequences exceeded the lower limit (default: 90%).
This means low quality sequence read. |
| |
unconv:
the number of unconverted CpHs (CpA, CpC and CpT) exceeded the upper limit (default: 5).
This means incomplete bisulfite conversion or low quality sequence read. |
| |
% conv: percent of "number of converted CpHs" / "number of CpHs" exceeded the lower limit (default 95%).
This means incomplete bisulfite conversion or low quality sequence read. |
| |
user desired: sequence was excluded by checking on the "exclude" checkbox. |
|
|